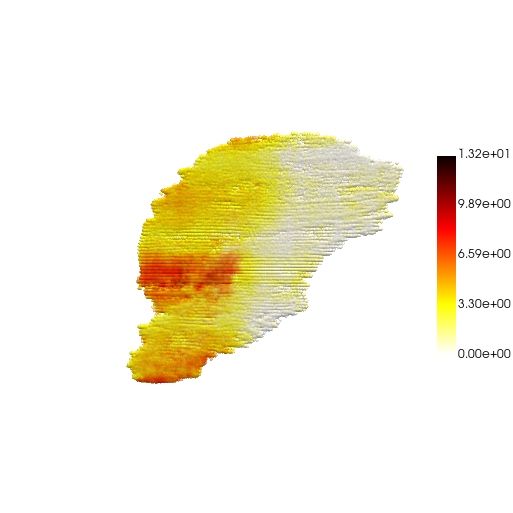
Gene Imputation (3D)
Note
The 3D Gene Imputation is computationally intensive and may require several hours to complete, depending on dataset size and hardware configuration.
!git clone https://github.com/lanshui98/UniST.git
%cd UniST
!pip install -r requirements.txt
Note that the spatial information should be at adata.obsm['spatial'].
Put the adata under external/SUICA_pro/data/.
%cd external/SUICA_pro
Read the Data
!pip install scanpy
import scanpy as sc
adata = sc.read('data/3D_data.h5ad')
genes = adata.var_names
Example: get the index of gene “Myl2”
gene_idx = genes.get_loc("Myl2")
print(gene_idx)
Visualization
from unist.downstream.vis import construct_pc, three_d_plot
# if you encounter: initialized module 'pyvista' has no attribute '_plot', try this:
!python scripts/patch_pyvista_circular_import.py
adata.obs["Myl2"] = adata.X[:,gene_idx].toarray().copy()
pc, cmap = construct_pc(
adata=adata,
spatial_key="spatial",
groupby="Myl2",
colormap="hot_r"
)
three_d_plot(
model=pc,
key="Myl2",
colormap="hot_r",
model_style="points",
model_size=4.0,
show_legend=True,
jupyter="trame",
legend_loc="center right",
opacity=0.5
)
"static"for static image"trame"for interactive window (need to installnest_asyncio2)
For more 3D visualization/animation details, please go to Animation.
Step1: Train GAE
! python train.py --mode embedder --conf ./configs/ST/embedder_gae_3d_sparse.yaml
Visualize the embeddings
emb = sc.read('logs/GAE-3D-sparse/3d_sparse/lightning_logs/version_0/embedded-all.h5ad')
emb.obs["emb1"] = emb.obsm["embeddings"][:, 0]
pc, cmap = construct_pc(
adata=emb,
spatial_key="spatial",
groupby="emb1",
colormap="viridis_r",
)
three_d_plot(
model=pc,
key="emb1",
colormap="viridis_r",
model_style="points",
model_size=4.0,
show_legend=True,
jupyter="trame",
legend_loc="center right",
opacity=0.5
)
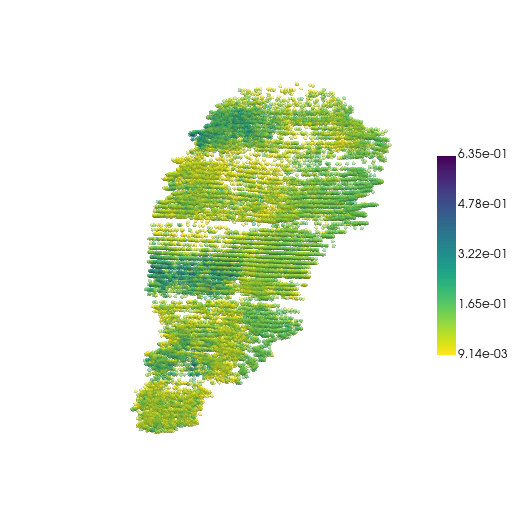
Notice how the z-axis is amplified here.
Certain parameters are set to handle sparse z-direction in 3D spatial transcriptomics data (e.g., when slice spacing is large).
use_anisotropic_knn: True
Meaning: Whether to use anisotropic KNN graph construction
z_weight: 2.0
Meaning: Weight factor for z-direction
Effect:
> 1: Reduces the influence of z-direction distanceCalculation:
weighted_z = z / z_weightFor example,
z_weight=2.0means z-direction distance is halved, making z-direction points more likely to become neighbors
Principle:
Original distance: d = sqrt((x1-x2)² + (y1-y2)² + (z1-z2)²) Weighted: d = sqrt((x1-x2)² + (y1-y2)² + (z1-z2)²/z_weight²)
z_threshold: null
Meaning: Maximum connection distance threshold in z-direction
Effect:
null/None: Automatically set to 30% of z-direction rangeNumeric value: Manually set maximum z-distance (in original coordinate units)
Principle:
Even after weighting, if two points are too far apart in z-direction, they should not be connected
Example:
# If z_range = 1000 (from z=0 to z=1000) # z_threshold = null → automatically set to 1000 * 0.3 = 300 # This means points with z-direction distance > 300 will not be connected
preserve_z_scale: True
Effect:
True: z-direction is not compressed, maintaining a relatively larger rangeFalse: z-direction is compressed together with xy-directions to the same range
Principle:
# preserve_z_scale = False # All directions compressed to [-1, 1], maintaining aspect ratio scale_x = x_range / max(x_range, y_range, z_range) scale_y = y_range / max(x_range, y_range, z_range) scale_z = z_range / max(x_range, y_range, z_range) # preserve_z_scale = True # z-direction maintains larger range, not compressed scale_x = x_range / max(x_range, y_range) scale_y = y_range / max(x_range, y_range) scale_z = z_scale_factor
z_scale_factor: 1.5
Meaning: Scaling factor for z-direction (only effective when
preserve_z_scale=True)Effect:
= 1.0: z-direction maintains original relative scale> 1.0: Amplifies z-direction importance (recommended for sparse z-direction)
Principle:
normalized_z = (z - z_min) / z_range # Normalize to [0,1] normalized_z = (normalized_z - 0.5) * 2.0 # Transform to [-1,1] normalized_z = normalized_z * z_scale_factor # Apply scaling factor
Step2: Train INR + fine-tune GAE
! python train.py --mode inr --conf ./configs/ST/inr_embd_3d_sparse.yaml
Fourier Feature Encoding Parameters for sparse z-direction:
encoding_scales: [1, 10, 100]
Meaning: Multi-scale frequency bands for Fourier feature encoding
Effect:
Each scale creates a separate frequency encoding
[1, 10, 100]means three frequency bands: low (1), medium (10), and high (100)Higher scales capture finer details, lower scales capture global patterns
Principle:
For each scale s in [1, 10, 100]: - Generate random matrix B with scale s - Encode: sin(2π * x @ B), cos(2π * x @ B) - Concatenate all encodings
Why multiple scales?
Single scale can only capture one frequency range
Multi-scale captures both global (scale=1) and local (scale=100) patterns
Better representation for complex spatial structures
anisotropic_3d: True
Meaning: Use different frequency encodings for xy-directions and z-direction
Effect:
True: xy-directions and z-direction are encoded separately with different frequenciesFalse: All directions use the same encoding frequencies
Why needed?
z-direction is sparse (large slice spacing)
z-direction needs lower frequencies to capture slice-level patterns
xy-directions need higher frequencies to capture within-slice details
Implementation (see
external/SUICA_pro/networks/ffn.py, lines 68-83):if anisotropic_3d: # xy-direction encoding (first 2 dimensions) xy_encodings = [GaussianEncoding(2, mapping_size, scale=s) for s in encoding_scales] # [1, 10, 100] # z-direction encoding (3rd dimension) z_encodings = [GaussianEncoding(1, mapping_size, scale=s) for s in z_scales] # [0.1, 1.0, 10.0]
z_scales: [0.1, 1.0, 10.0]
Meaning: Frequency scales specifically for z-direction encoding
Effect:
Only used when
anisotropic_3d=Truez-direction uses these scales instead of
encoding_scalesTypically lower than xy-direction scales (because z is sparse)
Comparison with
encoding_scales:xy-direction: encoding_scales = [1, 10, 100] (higher frequencies) z-direction: z_scales = [0.1, 1.0, 10.0] (lower frequencies) Ratio: z_scales are approximately 10x lower than encoding_scales → z-direction uses 10x lower frequencies → Captures slice-level patterns, not fine z-direction details
Why lower frequencies for z?
z-direction is sparse: large distances between slices
High frequencies would create noise between distant slices
Low frequencies capture smooth variations across slices
Step3: Prediction/Imputation
Prepare normalized custom coords
! python prepare_custom_coords.py --mode 3d --reference data/3D_data.h5ad --coords data/3D_coords.xyz --output data/preprocessed_data/custom_coords_3d_norm.npy --keep_ratio True --preserve_z_scale True --z_scale_factor 1.5
Run prediction
! python predict.py --mode inr --conf ./configs/ST/inr_pred_3d_sparse.yaml
Map reconstructed coords back to original space
! python map_coords_back.py --reconstructed reconstructed-custom-3d.h5ad --reference data/3D_data.h5ad --output reconstructed-original-3d.h5ad --mode 3d --keep_ratio True --preserve_z_scale True --z_scale_factor 1.5
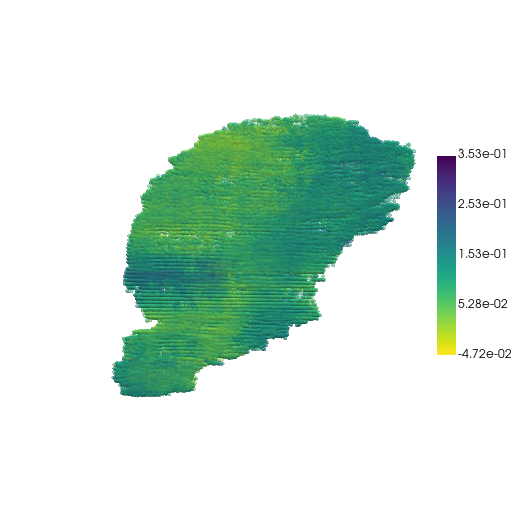
Visualize the fitted embeddings
res = sc.read("logs/GAE+FFN-3D-sparse/3d_sparse/lightning_logs/version_1/reconstructed-original-3d.h5ad")
res.obs["emb1"] = res.obsm["fitted_embd"][:, 0]
res.obs["emb1"] = res.obsm["fitted_embd"][:, 0].toarray().flatten()
pc, cmap = construct_pc(
adata=res,
spatial_key="spatial",
groupby="emb1",
colormap="viridis_r",
)
three_d_plot(
model=pc,
key="emb1",
colormap="viridis_r",
model_style="points",
model_size=4.0,
show_legend=True,
jupyter="trame",
legend_loc="center right",
opacity=0.5
)
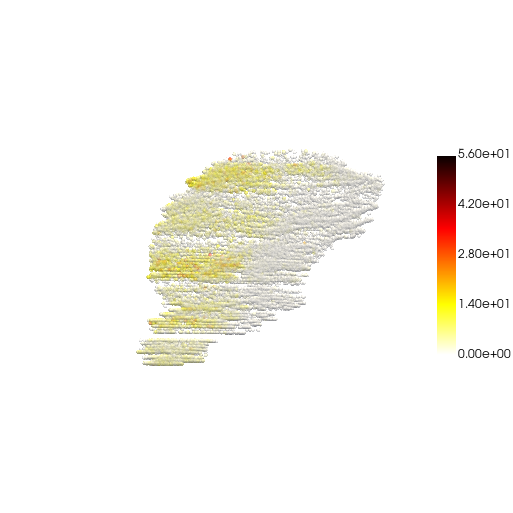
Visualize the result
res.obs["gene1"] = res.obsm["reconstructed_raw"][:, 0]
pc, cmap = construct_pc(
adata=res,
spatial_key="spatial",
groupby="gene1",
colormap="hot_r",
)
three_d_plot(
model=pc,
key="gene1",
colormap="hot_r",
model_style="points",
model_size=4.0,
show_legend=True,
jupyter="trame",
legend_loc="center right",
opacity=0.5
)